医療機器単一調査プログラム(MDSAP)は、オーストラリア、ブラジル、カナダ、日本、米国の各国における医療機器規制で要求されるQMS関連要求事項とISO13485:2016に基づき実施される品質マネジメントシステム監査を5ヵ国のメンバーにより構成されるQMS調査機関が一度の監査により調査を実施するプログラムです。MDSAPを活用する利点については、当社のMDSAPサービスのページをご覧ください。本記事では、MDSAPに基づく調査報告書 が発行されるまでに必要な主な手順について解説します。
MDSAPによる適合性調査報告書を取得するには、対象組織の品質マネジメントシステム(QMS)が少なくともISO 13485:2016規格の該当するすべての要求事項を満たしている必要があります。さらに、製造業者が医療機器を販売する、または販売を希望するMDSAP参加各国の医療機器規制に基づき求められるQMS要求事項への適合も必要となります。MDSAPは5つの参加国を対象としていますが、QMSについては製造業者が適用を受ける国の要求事項のみを満たせばよいとしています。そのため、製造業者が最初に米国とオーストラリアの2ヵ国で製品を販売するとした場合、QMSが初回監査において満たす必要があるのは、米国食品医薬品局(U.S. Food and Drug Administration: FDA)およびオーストラリア薬品・医薬品行政局(Australia Therapeutic Goods Administration: TGA)の該当する要求事項のみです。将来的に、MDSAPの他の参加国での販売を開始する場合は、追加する国の要求事項をQMSの範囲として広げていく必要があります。
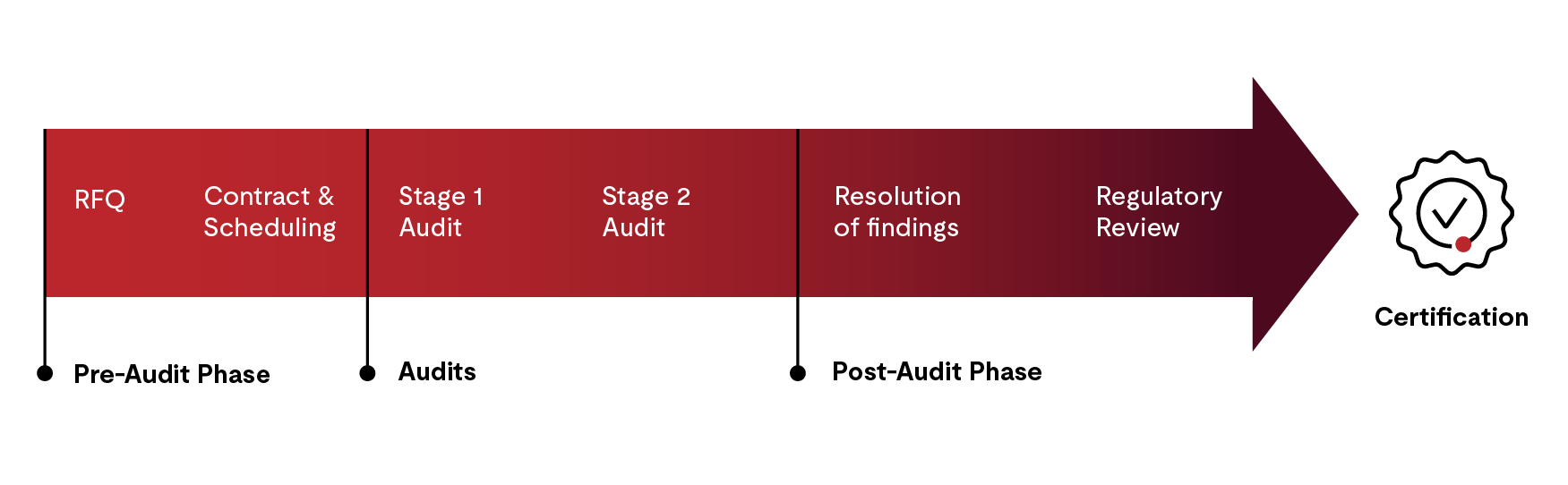
弊社が提供するMDSAPのプロセスには4つのステップがあります。
- 見積もり、契約、および監査スケジュール策定のための事前監査フェーズ
- 第1段階および第2段階の監査
- 監査後の活動
- QMSの調査報告書及び適合証の発行
各ステップを確実に進めるには、十分な準備とQMSの運用を継続する経営陣の強いコミットメントが必要になります。
事前監査フェーズ
契約に先立ち、お客様の組織体制や施設に関する基本情報、市場投入を予定している製品(医療機器)、従業員数などの詳細情報を提供いただく必要があります。UL Solutionsは、提供された情報に基づいた御見積を契約締結前に提示します。お客様より見積内容の承諾を得られた後、お客様とUL Solutionsの間で契約が締結され、調査日程が決定します。
第1段階の監査
第1段階の監査(S1)の主な目的は、お客様の品質マネジメントシステムがMDSAPの要求事項を満たすための準備が整っているかを評価することです。特に、ISO 13485 やQMS要求事項に沿って文書化された手順(SOPおよび関連文書)が充填的に確認されます。SOPには、ISO 13485:2016およびMDSAP監査アプローチ文書(MDSAP AU P0002)で定義されたすべての適用プロセスが網羅されていることが求められます。MDSAP監査アプローチ文書は、MDSAPに関連する文書としてウェブサイトで公開されています。ISO 13485:2016の要求事項およびMDSAPによる監査アプローチのガイドラインをよく理解し、組織の品質マネジメントシステムSに反映させる必要があります。第1段階の監査は、実地またはリモートで実施できます。すべてのSOPを電子化し、それらをリモートで閲覧できるようにしておくことも大事です。監査では、対象となる組織の品質マニュアルが詳細に精査されます。
MDSAP参加国の中には、海外の製造業者が最初の市場認可プロセスにおいて、現地代理人の支援を受ける必要がある国もあれば、任意とされている国もあります。
- オーストラリアでは、海外の製造業者はオーストラリアに拠点を持つ事業者を代理人として任命し、TGAに医療機器を登録する必要があります。この代理人は「Australian sponsor」と呼ばれます。
- ブラジルでは、海外の製造業者は製造販売の認可を取得するために、現地の事業者を「Brazilian registration holder (BRH)」として指定する必要があります。
- 日本においては、業許可をもつ製造販売業「marketing authorization holder(MAH:製造販売業者)」が厚生労働省とのやり取りを行う必要があります。
- カナダでクラスII以上の医療機器を販売するために製造業者がMDSAPにより適合認証を取得する必要があります。MDSAP参加国の中でMDSAPでの適合証明が義務付けられているのは、現在はカナダのみです。なお、Health CanadaのクラスIの医療機器については、MDSAPは求められていません。カナダでは、海外の製造業者がカナダ国内の輸入業者を任命することも可能ですが、必須ではありません。製造業者は Health Canadaに直接医療機器のライセンスを申請することができます。また、Health Canadaとのやり取りを支援する第三者の規制コンサルタントを活用することもできますが、義務ではありません。
- 米国では、医療機器製造業者は米国食品医薬品局(FDA)と直接やり取りすることができます。
第1段階の監査(S1)では、適用される各規制当局の基本要件について認識していることを示さなければなりません。S1監査で確認された指摘事項はお客様に通知され、第2段階の監査(S2)前に対応する必要があります。指摘事項の発見は、次の2つの結果につながる可能性があります。
- お客様の品質マネジメントシステムはMDSAPに対応する準備が整っていないと判断され、新たなS1監査の実施が推奨されます。
- 特定された指摘事項が比較的軽微であると判断される場合、次のステップとしてS2監査が推奨されます。求められる要求事項との相違の程度に応じて、S2監査はS1監査の1ヵ月後から6ヵ月後の間でスケジュールされ、間隔が6ヵ月を超える場合には、新たにS1監査を実施する必要があります。
第2段階の監査
第2段階の監査は現地で実施され、通常は数日間にわたります。調査日数は契約締結前に製造業者に通知されます。この監査は、MDSAPの監査アプローチモデルを使用して実施され、品質マネジメントシステムのさまざまな側面を評価するために、以下の構成が求められます。
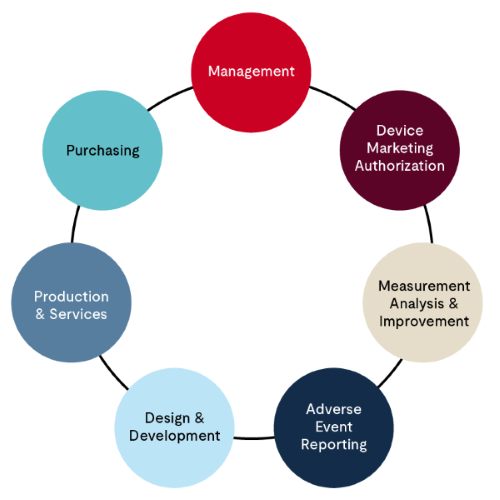
- 経営プロセス - このプロセスは11のタスクに分かれており、組織体制、経営層によるコミットメント、マネジメントレビュー、文書管理、人材、能力開発などを含みつつ、これらに限定されない品質マネジメントシステムの概要に焦点を当てます。品質マニュアルはこの評価の重要な要素です。
- 医療機器製造販売承認可プロセス - 適用される規制当局への施設登録および医療機器製造販売承認可プロセスに重点をおきます。これには3つのタスクがあり、これらのプロセスは、多くの場合、MDSAP認証を目指す組織内の規制の専門家によって管理されます。
- 測定、分析、および改善 - その名のとおり、品質マネジメントシステムの機能を監視し、改善に必要な取り組みを行います。苦情対応、CAPA、内部監査プロセス、非適合の材料とプロセス、市販後監視がこのプロセスの中核となります。これらのプロセスは、多くの場合、組織内の品質保証部門のマネージャーによって管理されます。
- 有害事象の報告、リコールおよび勧告通知 - このプロセスには、MDSAPの監査アプローチの下で評価される3つのタスクがあります。これらのプロセスは、多くの場合、組織内の薬事部門によって管理されます。
- 設計および開発プロセス - MDSAPの監査アプローチでは、新しい機器を設計・開発する際の計画から設計移管、製造までを対象とする16のタスクがあります。設計変更プロセスもこれに含まれます。
- 生産およびサービス管理プロセス - MDSAPの監査アプローチでは、生産およびサービス管理プロセスを対象とする29のタスクが推奨されています。このセクションは、インフラ、作業環境、製造プロセスの妥当性確認、製造品質管理試験、保管、識別、トレーサビリティ、非適合材料の取り扱い、販売など、医療機器の製造に関わるすべてを網羅しています。
監査後の活動
監査後の活動には、主に2つの活動が含まれます。
- 監査中に特定された不適合に対する製造業者の対応
- 監査機関による監査結果に関する薬事対応部門内部規制レビュー
S1監査で特定された不適合は、S2監査で対処、除去、検証する必要があります。S2監査で不適合が特定された場合、MDSAPの要求事項に従って、1から5の等級に評価されます。この評価基準の基本概念は、MDSAPがGHTF/SG3/N19:2012から採用したものです。このGHTFの原文文書はISO 13485:2016よりも前に作成されたものです。そのため、文書全体にISO 13485:2003への言及が見られますが、記載されている評価基準の原則と概念は引き続き適用されます。等級4および5の不適合、または ISO 13485 の「重大な指摘事項」と評価された不適合は、MDSAP調査に基づく適合証明書が発行される前に解決する必要があります。
監査チームは監査結果を内部規制レビューチームに提出します。このレビューの結果に基づいて、認証決定者は、MDSAPに基づく認証書を発行するのに十分な調査結果かどうかを判断し、決定事項を製造業者に通知します。最終報告書パッケージは、MDSAPの方針に従って、MDSAP REPSデータベースにも登録されます。
MDSAPによる適合証明書は、MDSAP AU P0026の要求事項に従う必要があり、最低限、法的製造業者の名称と住所、適用範囲、管轄地域、品質マネジメントシステムの運用開始日、有効期限、および適合証明書の有効期限が記載されます。通常、適合証明書は3年間有効です。
MDSAPにおけるUL Solutionsの特徴
UL Solutionsは、安全科学分野の世界的なリーダーです。ISO 17021の英国認証機関認定審議会(UKAS)認定登録機関として、複数のプログラムのQMS登録を提供するとともに、ISO 13485の監査やISO 13485とISO 9001の統合監査も提供しています。
調査を実施するすべての監査員は、UL Solutionsの正社員であり、お客様の品質マネジメントシステムのライフサイクルを通じて一貫した監査とサポートを提供します。
当社の経験豊富な監査員や評価員は、40か国以上で働く15,000人の使命感あふれる従業員の一員として活動し、100か国以上のお客様にサービスを提供しています。
ULの認証マークは、信頼性の証として世界的に認知されており、安全性に関する我々のミッションを推進するという揺るぎないコミットメントを反映しています。
執筆者について
Chira Dekaは、UL SolutionsのMDSAPのプログラムマネージャーとして勤務。ISO 13485、ISO 9001、MDSAPにおいて、主任審査員、認証決定者を務めるとともに、医療機器規則(MDR)および体外診断用機器規則(IVDR)の監査プログラムの主任監査員、UKCA認証プログラムの監査員でもある。Chiraは体外診断用医療機器(IVD)技術の技術専門家であり、この分野で複数の特許を取得。以前は、米国国立衛生研究所(NIH)の中小企業技術革新制度(SBIR)技術審査委員会(微生物学)のメンバーを務め、NIHおよび米国商務省による先端技術プログラム(ATP)研究のIVDおよびバイオテクノロジー助成金を受けた実績がある。




X
お気軽にお問い合わせください
弊社のサービスにご関心をお寄せいただき、ありがとうございます。お問合せ内容とお客様情報をご入力ください。後ほど担当者よりご連絡いたします。
Please wait…
残り文字数